Online GESS Tool
(Version 1.1.3) details
BACKGROUND
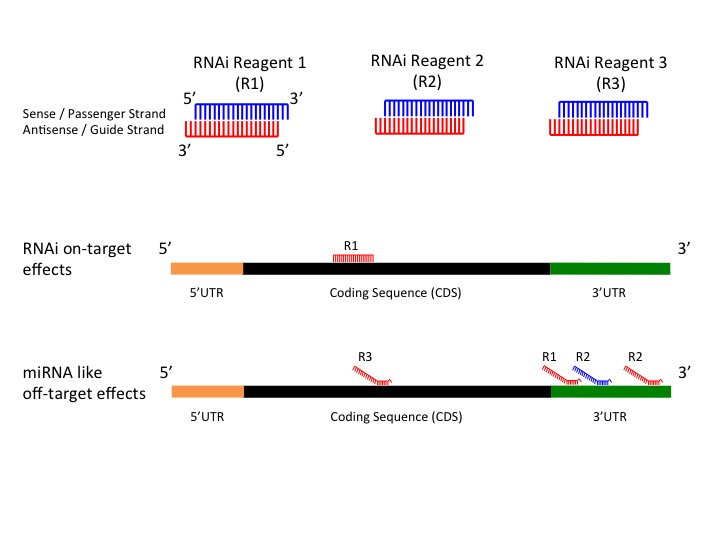
RNAi is a widely used and valuable genetics tool to study gene function, but it is vulnerable to some off-target effects representing a significant source of false positive results. Previously a bioinformatics method, genome-wide enrichment of seed sequence matches (GESS), was developed to identify candidate off-targetted transcripts from primary screen results. The algorithm reveals microRNA (miRNA)-like effects by seed region analysis. For more information please see A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. GESS online tool was developed to provide a quick and an easy to use user interface to run the GESS algorithm.
Figure 1: RNAi On-Target Effects versus RNAi miRNA like Off-Target Effects
GUIDE TO USE ONLINE GESS TOOL
Part 1: User Input
1- Upload si/shRNA file :
The user should upload a tab or comma separated text file or Excel file containing si/shRNA data. If both active and inactive si/shRNAs are provided, the input file should contain three columns; first column with si/shRNA identifiers, second column with si/shRNA sequences and the third column with corresponding phenotype information. If only active si/shRNAs are provided, the input file should have two columns; one for si/shRNA identifiers and one for the sequences. No phenotype data is needed in this case.
Case 1: Input file contains both active and inactive si/shRNAsIf a user is providing both active and inactive si/shRNAs, she/he has to submit a file containing at least three columns in the following order; a sequence identifier, si/shRNA sequence and corresponding phenotype information. Please see below for accepted input for each column.
|
Any identifier given by the user
For example: sequence_1, D-200-200, etc. |
RNAi reagent sequence, can be sense strand or antisense strand
For example: tttgggcatccgcctgtaaa , CGACAGAAGCAUUCCCUAU, etc. |
Phenotype data to distiguish "active" and "inactive" RNAi
To indicate active RNAi: "YES", "TRUE", 1, (or any number equal to or greater than 1) To indicate inactive RNAi: "NO", "FALSE", 0, (or any number smaller than 1) |
| 1 | GCAGCTTCATAACCGAAGA | Yes |
| 2 | GAGCAGCCCTTTAAGGATT | Yes |
| 3 | GAGCAGCCCTGGAAGGAC | No |
In this case the program will assume that the user provides only active si/shRNAs in the input file and it will generate a set of theoretical inactive si/shRNA seed sequences. To create an inactive set, the last nucleotide of each seed sequence will be changed to its compliment. As a result, there will be equal number of active si/shRNA seed sequences and inactive si/shRNA seed sequences to analyze.
A sample layout for input file without any phenotype/activity data can be seen below. Please see above to see accepted input formats.
| 1 | GCAGCTTCATAACCGAAGA |
| 2 | GAGCAGCCCTTTAAGGATT |
| 3 | GAGCAGCCCTGGAAGGAC |
Indicate Input Strand: Users have to state whether their RNAi reagent sequences are sense or antisense strands.
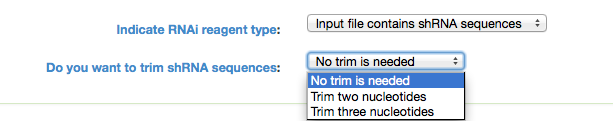
Indicate RNAi Reagent Type: GESS analysis can be done using siRNA or shRNA sequences. If the input contains shRNA seqeunces, it is possible to trim them by two or three nucleotides. If shRNA is selected another option pops up in the user interface (see below image) to make sure if the user wants to trim shRNA sequences or not. If one of the trim options is selected, the program removes the required number (chosen by user, two or three) of nucleotides from shRNA sequences.
Figure 2: shRNA Trimming Options
2- Choose Reference Data Types or Upload a Custom Database file:
The user can either choose to use the built-in database; available for human, mouse and fly; or provide a custom database file.
If the user wants to use the built-in database, she/he can choose the organism (Human Mouse or Fly) and transcript region (3’UTR, 5’UTR, CDS, Full Transcript for Protein Coding Genes, Full Transcript for All Genes) to search for the seed matches. The default values are “Human” for organism and “3'UTR” for region.
If the user wants to upload a custom database file, she/he can do the GESS analysis for any organism of interest. The file should have FASTA formatted sequences. A sample file can be seen here.
3- Options:
The user can change the parameters of the program and make it more or less stringent.
-
Seed Sequence Length:
The length of the seed sequences to be used in the GESS analysis. Seed sequences are extracted from si/shRNA sequences based on this parameter. The default value is 7. The user can choose 6 to make the search for sequence match less stringent or 8 to make it more stringent.
-
Minimum Number of Seed Matches:
The minimum number of seed matches to be found in the transcript sequence to consider a “match” between seed sequence and transcript sequence. The default is “1”, the user can ascend the number up to "4" to make the search more stringent.
-
Strand to use:
Figure 3: RNAi Seed Region Matches
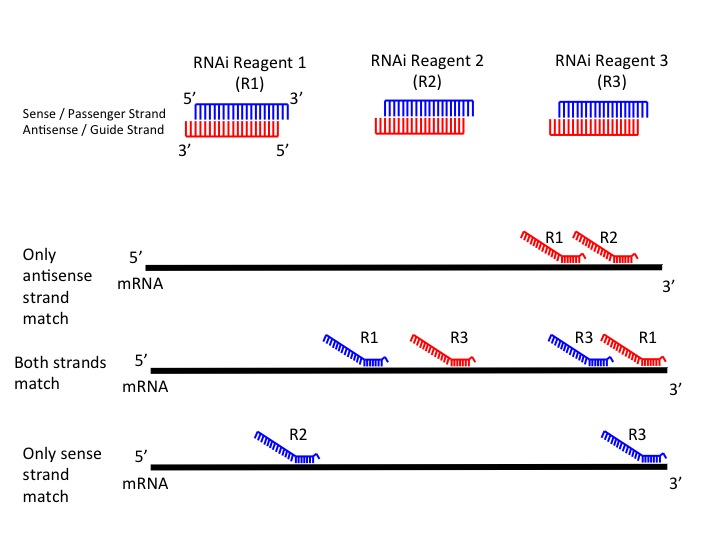
The program extracts antisense and sense strand seed sequences for each si/shRNA sequence and searches for matches in the transcript sequences and counts the number of seed matches between each seed sequence and transcript. If the number of seed matches is equal to or greater than the minimum number of seed matches selected by the user, the program will consider it a match between the si/shRNA and the tested transcript sequence.
Antisense (Guide) Strand Only: The program will use only antisense strand seed sequences while searching for the matches (Figure 1). This is the default value for the GESS analysis, the user can change this parameter to either of the following options.
Either Strand: The program will use both antisense and sense strand seed sequences while searching for the matches. It will count the number of matches for both cases and sum them. If the sum is greater than the minimum number of seed matches provided by the user, it will be considered a match.
Both Strands: The program will use both antisense and sense strand seed sequences as it does for "Either Strand" case, the only difference is that both antisense and sense strand seed sequences has to match to the tested sequence at least once.
Sense (Passenger) Strand Only: The program will only use sense strand seed sequences while searching for the matches.
-
p Value:
Significance threshold parameter to be used at the multiple testing correction algorithms. The default is 0.05, the user can make it more or less stringent.
The null hypothesis (there is no difference between the frequency of si/shRNAs with phenotype and si/shRNAs without phenotype having a seed match to a given sequence) is rejected if the p value calculated is less than the corrected p value threshold. p value given by the user (default is 0.05) is used to calculate corrected p value thresholds.
Three multiple hypothesis testing correction methods have been implemented in GESS; the Benforrini, the Benforrini step-down (Holm), the Benjamini and Hochberg False Discovery Rate correction (Simes') methods. The methods are listed below from the most stringent to least stringent one.
- The Bonferroni correction method: (α / A)
- The Bonferroni step-down (Holm) correction method: (α / (A + 1 - rank of sequence)
- The Benjamini and Hochberg False Discovery Rate correction method: (α * rank of sequence / A)
α is set to 0.05 by default and can be changed by the user. A is the number of transcript sequences analyzed in the study. The rank of the sequence is determined by sorting all analysed sequences according their p values.
4- Advanced Options:
-
Which transcripts do you want to be listed in the results file:
By default, all tested transcripts are listed in the results file but a user can get the results for only significant transcripts based on user selected the p value threshold by choosing “Only Significant Transcripts”.
-
Would you like to include transcripts that are enriched in the background to the results file:
By default, transcripts that are enriched among active RNAi reagents are listed in the results file. If "Yes" option is selected transcripts enriched among inactive RNAi reagents are also listed in the results file.
-
Would you like to scramble all si/shRNA seed sequences to run a control test?
The user can run a control test by scrambling all si/shRNA seed sequences. If "Yes" was selected, the program will randomly scramble each seed sequence and run a GESS analysis.
5- Job Id:
Providing a job identifier for a GESS analysis is optional; if it is available, the resulting files will be named according to it. Otherwise, the program will randomly create an identifier for the analysis.
6- Email:
The user should provide an email address in order to get the GESS analysis results. If the analysis results significant outcome, two text files will be sent to the user.
Part 2: Results
1- File with GESS analysis results:
This file contains the basic GESS analysis results. A sample file can be seen here. Each tested sequence is listed in this file in a line with the corresponding GESS analysis results. Detailed explanation of GESS analysis results table can ben found below.
|
Identifier for the tested sequence. If the built in database was used, version number is used in here. |
|
|
Gene symbol for mouse and human, FlyBase Identifier for fly. If custom database is provided, it will be empty. |
|
|
Tested transcripts are ranked from the one with lowest P value (rank=1) to the one with highest P value (rank = A, the number of sequences tested.) |
|
|
Seed Match Frequency of the active si/shRNAs |
|
|
Seed Match Frequency of the inactive si/shRNAs |
|
|
Seed Match Enrichment (Seed Match Frequency of the active si/shRNAs / Seed Match Frequency of the inactive si/shRNAs ) |
|
|
Active RNAi: Enrichment is among active RNAi reagents Inactive RNAi: Enrichment is among inactive RNAi reagents |
|
|
Number of active si/shRNAs that have seed matches to the tested sequence |
|
|
Number of active si/shRNAs that do NOT have seed matches to the tested sequence |
|
|
Number of inactive si/shRNAs that have seed matches to the tested sequence |
|
|
Number of inactive si/shRNAs that do NOT have seed matches to tested sequence |
|
|
If one of the si/shRNA categories (siPhenMatch, siPhenNoMatch, siNoPhenMatch, siNoPhenNoMatch) has 20 events or less, the FisherExactTest p-value will be used instead of the Yates Chi Square p-value. |
|
|
Either Yates Chi-Square p-value or Fisher Exact Test p-value depending on the p-value selected method. |
|
|
p-value adjusted according to Bonferroni correction method. |
|
|
p-value adjusted according to Bonferroni Step-down correction method. |
|
|
p-value adjusted according to Benjamini & Hochberg correction method. |
|
|
"Yes" if statistically significant according to Bonferroni correction method, "No" otherwise. |
|
|
"Yes" if statistically significant according to Bonferroni Step-down correction method, "No" otherwise. |
|
|
"Yes" if statistically significant according to Benjamini and Hochberg correction method, "No" otherwise. |
|
|
α / A |
|
|
α / (A + 1 - rank of sequence) |
|
|
α * rank of sequence / A |
2- File with tested sequences and matching si/shRNAs:
This file contains tested sequences and identifiers of matching "active" si/shRNAs. Tested sequences and matching active si/shRNAs are mapped in a line in this file. Sequence identifiers are ordered according to their ranks in the results file and only the significant results are reported here (p value cut-off 0.05). A sample file can be seen here.
Error Handling
1- Errors Detected While Pre-processing the Input si/shRNA File :
GESS program pre-processes input si/shRNA file and if detects errors in more than 25% of the si/shRNA data, it displays a warning message on the screen. In this case GESS analysis would not been started, users have to fix the errors in their input file. Error type and the row numbers with invalid data are listed on the UI. Users can see the content of each row by hovering their mouse on the little box with row number. Please see the screen shot below.
Figure 4: Error Page
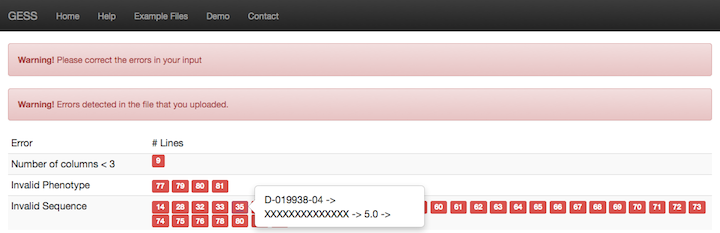
If the error rate is less than or equal to 25% in the input file, the program ignores the invalid data and does the analysis using the valid data only. When the analysis is completed, the user is informed about ignored si/shRNA sequences via email.
2- Errors Detected after GESS analysis started
If the GESS analysis fails for a reason after successfully submitting an input to the tool, an email is sent to user to inform him/her about the failure.
Version Details
Version 1.1.3 - May 2021- Fixed problem fly input file parsing, due to format change
- Fixed problem input file parsing would cause crash.
- Fixed problem where it would indicate finished when error experienced.
- Fixed problem with spaces in Job Name
Version 1.1
- Updated so email not used.
- Updated so all results results are zipped and moved to directory.
- Added link to page so users can come back to get results.